If we want to understand how the brain creates memories, and how genetic disorders distort the brain’s machinery, then the fragile X gene is an ideal place to start. That’s why the Stephen T. Warren Memorial Symposium, taking place November 28-29 at Emory, will be a significant event for those interested in neuroscience and genetics.
Stephen T. Warren, 1953-2021
Warren, the founding chair of Emory’s Department of Human Genetics, led an international team that discovered Read more
At a time when COVID-19 appears to be receding in much of Georgia, it’s worth revisiting the start of the pandemic in early 2020. Emory virologist Anne Piantadosi and colleagues have a paper in Viral Evolution on the earliest SARS-CoV-2 genetic sequences detected in Georgia.
Analyzing relationships between those virus sequences and samples from other states and countries can give us an idea about where the first COVID-19 infections in Georgia came from. We can draw Read more
In African Americans, the genetic risk landscape for inflammatory bowel disease (IBD) is very different from that of people with European ancestry, according to results of the first whole-genome study of IBD in African Americans. The authors say that future clinical research on IBD needs to take ancestry into account.
Findings of the multi-center study, which analyzed the whole genomes of more than 1,700 affected individuals with Crohn’s disease and ulcerative colitis and more than 1,600 controls, were published on February 17 in the American Journal of Human Genetics.
As part of their analysis, the researchers developed an algorithm that corrects for ancestry when calculating an IBD polygenic risk score. Polygenic risk scores are tools for calculating gene-based risk for a disease, which are used for IBD as well as other complex conditions such as coronary artery disease.
“Even though the disease destination looks the same, the populations look very different, in terms of what specific genes contribute to risk for IBD,” says lead author Subra Kugathasan, MD. “It shows that you can’t develop a polygenic risk score based on one population and apply it to another.”
Kugathasan is scientific director of the pediatric IBD program and director of the Children’s Center for Transplantation and Immune-mediated Disorders at Children’s Healthcare of Atlanta, as well as Marcus professor of pediatrics and human genetics at Emory University School of Medicine.
The first author of the paper is geneticist Hari Somineni, PhD, who earned his doctorate working with Kugathasan at Emory, and is now working at Goldfinch Bio in Massachusetts.
The primary sites to recruit study participants were Emory, Cedars-Sinai and Rutgers, along with Johns Hopkins and Washington University at Saint Louis. Along with Kugathasan, the co-senior authors and co-organizers of the study were Steven Brant, MD from Rutgers and Dermot McGovern, MD, PhD from Cedars-Sinai.
“One of our goals in treating IBD is to move toward a more personalized approach,” says McGovern, the Joshua L. and Lisa Z. Greer Chair in Inflammatory Bowel Disease Genetics at Cedars-Sinai. “Deciphering the genetic architecture is an important part of this effort. Studies such as this one are vital to ensure that diverse populations, including African-Americans, benefit from the tremendous advances promised by genomic medicine.”
As part of an effort to strengthen genomic surveillance for emerging strains of SARS-CoV-2, the Centers for Disease Control and Prevention (CDC) has awarded a contract to Emory University researchers to characterize viral variants circulating in Georgia.
The two-year contract is part of the SPHERES (SARS-CoV-2 Sequencing for Public Health Emergency Response, Epidemiology and Surveillance) initiative, with roughly $620,000 in total costs. The principal investigator is Anne Piantadosi, MD, PhD, assistant professor of pathology and laboratory medicine, with co-investigator Mehul Suthar, PhD, assistant professor of pediatrics (infectious diseases).
Both Piantadosi and Suthar are affiliated with Emory University School of Medicine and Emory Vaccine Center. Additional Emory partners include assistant professor of medicine Ahmed Babiker, MBBS, assistant professor of medicine Jesse Waggoner, MD and assistant professor of biology Katia Koelle, PhD.
“We are analyzing SARS-CoV-2 genomes from patients in Georgia to understand the timing and source of virus introduction into our community,” Piantadosi says. “We want to know whether there have been population-level changes in the rates of viral spread, and whether there are associations between viral genotype, viral phenotype in vitro, and clinical phenotype or clinical outcome.”
Supported by a $8 million, five-year grant, an Emory-led team of scientists plans to investigate new therapeutic approaches to fragile X syndrome, the most common inherited intellectual disability and a major single-gene cause of autism.
Fragile X research represents a doorway to a better understanding of autism, and learning and memory. The field has made strides in recent years. Researchers have a good understanding of the functions of the FMR1 gene, which is silenced in fragile X syndrome.
Still, clinical trials based on that understanding have been unsuccessful, highlighting limitations of current mouse models. Researchers say the answer is to use “organoid” cultures that mimic the developing human brain.
The new grant continues support for the Emory Fragile X Center, first funded by the National Institutes of Health in 1997. The Center’s research program includes scientists from Emory as well as Stanford, New York University, Penn and the University of Southern California. The Emory Center will be one of three funded by the National Institutes of Health; the others are at Baylor College of Medicine and Cincinnati Children’s Hospital Medical Center.
The co-directors for the Emory Fragile X Center are Peng Jin, PhD, chair of human genetics, and Stephen Warren, PhD, William Patterson Timmie professor and chair emeritus of human genetics. In the 1980s and 1990s, Warren led an international team that discovered the FMR1 gene and the mechanism of trinucleotide repeat expansion that silences the gene. This explained fragile X syndrome’s distinctive inheritance pattern, first identified by Emory geneticist Stephanie Sherman, PhD.
“Fragile X research is a consistent strength for Emory, stretching across several departments, based on groundbreaking work from Steve and Stephanie,” Jin says. “Now we have an opportunity to apply the knowledge we and our colleagues have gained to test the next generation of treatments.”
Fragile X researchers from three Emory departments, following COVID-19 spacing guidelines in the laboratory. From left to right: Peng Jin, Gary Bassell, Zhexing Wen and Nisha Raj.
Looking ahead, a key element of the Center’s research will involve studying the human brain in “disease in a dish” models, says Gary Bassell, PhD, chair of cell biology. Nisha Raj, PhD, a postdoctoral fellow in Bassell’s lab, has been studying how FMR1 regulates localized protein synthesis at the brain’s synapses.
“What we’re learning is that there may be different RNA targets in human and mouse cells,” he says. “There’s a clear need to regroup and incorporate human cells into the research.”
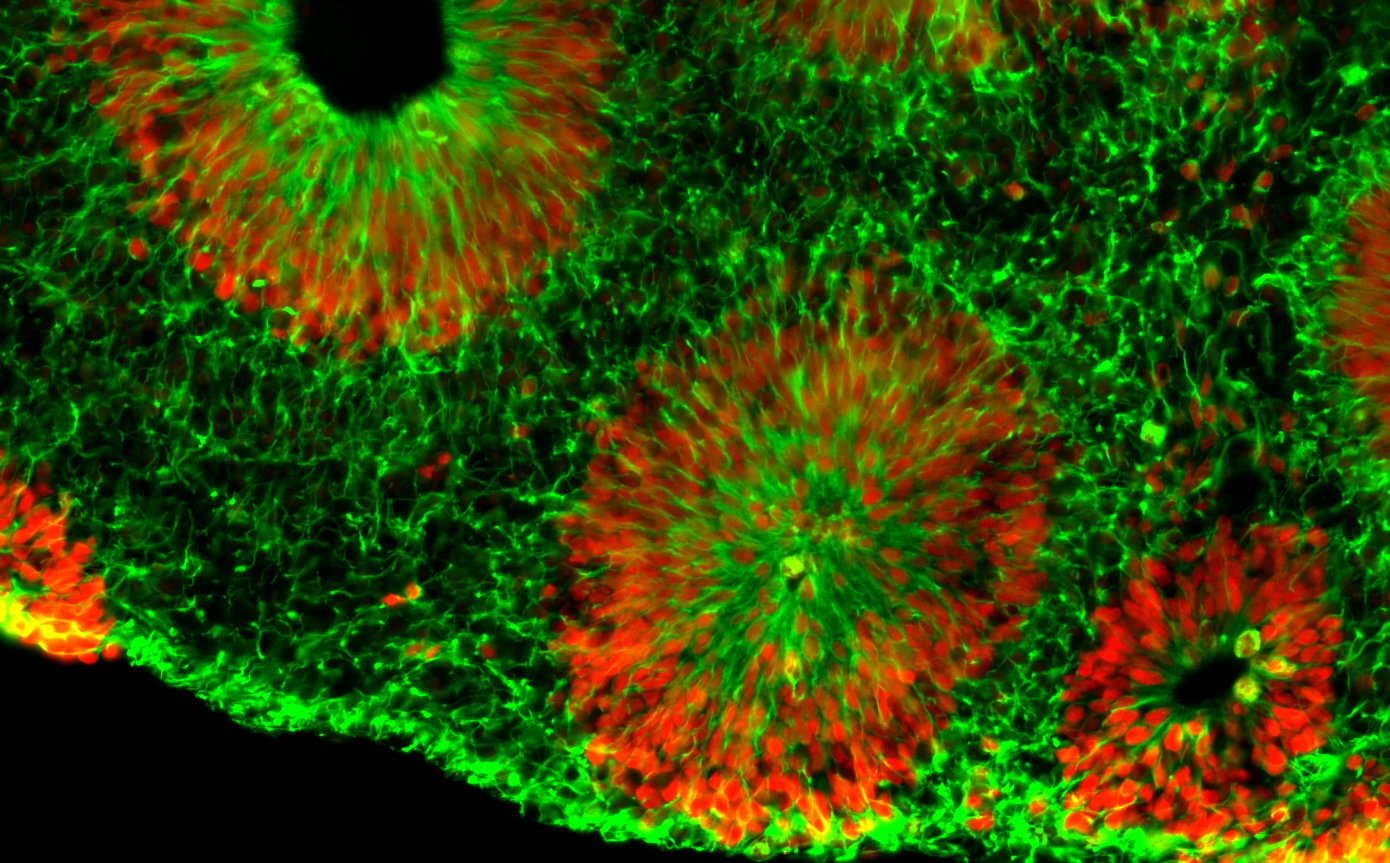
Microscope image of fragile X human brain organoids, courtesy of Zhexing Wen. Green represents cytoplasmic Nestin while red represents nuclear Sox2; both are markers for neural progenitor cells.
Center investigator Zhexing Wen, PhD, has developed techniques for culturing brain organoids (image above), which reproduce features of human brain development in miniature. Wen, assistant professor of psychiatry and behavioral sciences, cell biology and neurology at Emory, has used organoids to model other disorders, such as schizophrenia and Alzheimer’s disease.
The organoids are formed from human brain cells, coming from induced pluripotent stem cells, which are in turn derived from patient-donated tissues. Emory’s Laboratory of Translational Cell Biology, directed by Bassell, has developed several lines of induced pluripotent stem cells from fragile X syndrome patients.
“All of the investigators are sharing these valuable resources and collaborating on multiple projects,” Bassell says.
Principal investigators in the Emory Fragile X Center are Jin, Warren, Bassell, and Wen, along with Eric Klann, PhD at New York University, Lu Chen, PhD, and 2013 Nobel Prize winner Thomas Südhof, MD. Chen and Südhof are neuroscientists at Stanford.
Co-investigators include biostatistician Hao Wu, PhD and geneticist Emily Allen, PhD at Emory, neuroscientist Guo-li Ming, MD, PhD, at University of Pennsylvania, and biomedical engineer Dong Song, PhD, at University of Southern California.
Allen, Warren and Jin are part of an additional grant to Baylor, Emory and University of Michigan investigators, who are focusing on FXTAS (fragile X-associated tremor-ataxia syndrome) and FXPOI (fragile X-associated primary ovarian insufficiency). These are conditions that affect people with fragile X premutations.
Fragile X syndrome is caused by a genetic duplication on the X chromosome, a “triplet repeat” in which a portion of the gene (CGG) gets repeated again and again. Fragile X syndrome affects about one child in 5,000, and is more common and more severe in boys. It often causes mild to moderate intellectual disabilities as well as behavioral and learning challenges. About a third of children affected have characteristics of autism, such as problems with eye contact, social anxiety, and delayed speech.
The award for the Emory Fragile X Center is administered by the Eunice Kennedy Shriver National Institute of Child Health and Human Development, with funding from the National Institute of Mental Health and the National Institute of Neurological Disorders and Stroke.
In case you missed it, the 2017 Nobel Prize in Medicine marked the arrival of the flourishing circadian rhythm field. Emory Eye Center’s Mike Iuvone teamed up with Gianluca Tosini at Morehouse School of Medicine to probe how a genetic disruption of circadian rhythms affects the retina in mice.
Removal of the Bmal1 gene – an essential part of the body’s internal clock — from the retina in mice was known to disrupt the electrical response to light in the eye. The “master clock” in the body is set by the suprachiasmatic nucleus, part of the hypothalamus, which receives signals from the retina. Peripheral tissues, such as the liver and muscles, have their own clocks. The retina is not so peripheral to circadian rhythm, but its cellular clocks are important too.
What the new paper in PNAS shows is that removal of Bmal1 from the retina accelerates the deterioration of vision that comes with aging, but it also shows developmental effects – see below.
You might think: “OK, the mice have disrupted circadian rhythms for their whole lives, so that’s why their retinas are messed up.” But the Emory/Morehouse experimenters removed the Bmal1 gene from the retina only.
P. Michael Iuvone, PhD, director of vision research at Emory Eye Center
The authors write: “BMAL1 appears to play important roles in both cone development and cone viability during aging… Cones are known to be among the cells with highest metabolism within the body and therefore, alteration of metabolic processes within these cells is likely to affect their health status and viability.”
…Bmal1 removal significantly affects visual information processing and reduces the thickness of inner retinal layers. The absence of Bmal1 also affected visual acuity and contrast sensitivity. Another important finding was a significant age-related decrease in the number of cone photoreceptors (outer segments and nuclei) in mice lacking Bmal1, which suggests that these cells are directly affected by Bmal1 removal.
“When we genetically disrupted the circadian clocks in the retinas of mice, we found accelerated age-related cone photoreceptor death, similar to that in age-related macular degeneration in humans,” Iuvone says. “This loss of photoreceptor cones affects retinal responses to bright light.
“We also noted developmental effects in young mice,” Iuvone continues, “including abnormalities in rod bipolar cells that affected dim light responses. These findings have potential implications for pregnant shift workers and other women with sleep and circadian disorders, whose offspring might develop visual problems due to their mother’s circadian disruption.”
A marathon sleeper who got away left some clues for Emory and University of Florida scientists to follow. What they found could provide benefits for patients with the genetic disease myotonic dystrophy (DM) and possibly the sleep disorder idiopathic hypersomnia (IH).
The classic symptom for DM is: someone has trouble releasing their grip on a doorknob. However, the disease does not only affect the muscles. Clinicians have recognized for years that DM can result in disabling daytime sleepiness and sometimes cognitive impairments. At the Myotonic Dystrophy Foundation meeting in September, a session was held gathering patient input on central nervous system (CNS) symptoms, so that future clinical trials could track those symptoms more rigorously.
Emory scientists are investigating this aspect of DM. Cell biology chair Gary Bassell was interested in the disease, because it’s a triplet repeat disorder, similar to fragile X syndrome, yet the CNS mechanisms and symptoms are very different. In DM, an expanded triplet or quadruplet repeat produces toxic RNA, which disrupts the process of RNA splicing, affecting multiple cell types and tissues.
Rye at San Francisco myotonic dystrophy meeting. Photo courtesy of Hypersomnia Foundation.
Neurologist and sleep specialist David Rye also has become involved. Recall Rye’s 2012 paper in Science Translational Medicine, which described a still-mysterious GABA-enhancing substance present in the spinal fluid of some super-sleepy patients. (GABA is a neurotransmitter important for regulating sleep.)
In seven of those patients, his team tested the “wake up” effects of flumazenil, conventionally used as an antidote to benzodiazepines. One of those patients was an Atlanta lawyer, whose recovery was later featured in the Wall Street Journal and on the Today Show. It turns out that another one of the seven, whose alertness increased in response to flumazenil, has DM.
In an overnight sleep exam, this man slept for 12 hours straight – the longest of the seven. But an IH diagnosis didn’t fit, because in the standard “take a nap five times” test, he didn’t doze off very quickly. He became frustrated with the stimulants he was given and sought treatment elsewhere, Rye says. Lab Land doesn’t have all the details of this patient’s history, but eventually he was diagnosed with DM, which clarified his situation. Read more
In a study published this month in Hepatology, a multinational team of researchers describes a newly identified cause of congenital diarrhea and liver disease in children.
The rare disorder is characterized by significant diarrhea beginning soon after birth, low serum levels of fat-soluble vitamins and evidence of liver disease. Despite continued symptoms, with medical support, the children grow and develop normally, at least to the age of 12.
From left to right: Mutaz Sultan, Orly Elpeleg and Paul Dawson, representing three collaborating institutions.
Researchers from Emory University School of Medicine and Children’s Healthcare of Atlanta, working with colleagues from Makassed Hospital, Al-Quds University and Hadassah Medical Center, Hebrew University of Jerusalem studied a family with two children from the Palestinian territories who suffer from the disorder.
The team found that both children had inherited a mutation in a gene responsible for the transport of bile acids, which facilitate the digestion and absorption of dietary fats and fat-soluble vitamins. Although mutations had been identified in other genes important for the recycling of bile acids, this is the first report in humans of disease-associated defects in this gene, called Organic Solute Transporter-beta (SLC51B).
Almost 20 years ago, pediatric GI & hepatology researcher Paul Dawson, PhD, and colleagues identified mutations in another bile acid transporter gene (ASBT; SLC10A2) that caused congenital bile acid diarrhea.
“Even at that time, we knew that there were patients with similar symptoms that did not carry mutations in ASBT. But the genetic cause remained a mystery.” Dawson says. “What’s distinctive about this report is that these patients also have features of liver disease, which was not observed in previously described congenital bile acid diarrhea patients.” Read more
Kristen Thomas, PhD, now a postdoctoral fellow at St Jude Children’s Research Hospital
Schizophrenia genetics and its complexities are beginning to yield to large genome-wide studies. One of the recently identified top risk loci, miR 137, can be seen as a master key that unlocks other doors. The Mir 137 locus encodes a micro RNA that regulated hundreds of other genes, and several of those are also linked to schizophrenia.
Earlier this month, Emory’s chair of cell biology Gary Bassell and former graduate student Kristen Thomas published a paper in Cell Reports analyzing how perturbing Mir 137 affects signaling in neurons. Inhibiting Mir 137 blocked neurons’ responses to neuregulin and BDNF, well-known growth factors.
“We think a particularly interesting aspect of our paper is that it links miR137, neuregulin and ErbB4 receptor: three molecules with known genetic risk for schizophrenia,” Bassell writes. Read more
NMDA receptors are complex electrochemical machines, important for signaling between brain cells. Rare mutations in the corresponding genes cause epilepsy and intellectual disability.
Pre-M1 helices in multi-subunit NMDA receptor. Adapted from Ogden et al PLOS Genetics (2017).
In Emory’s Department of Pharmacology, the Traynelis and Yuan labs have been harvesting the vast amounts of information now available from public genome databases, to better understand how changes in the NMDA receptor genes relate to function. (Take a “deeper dive” into their November 2016 publication on this topic here.)
Their recent paper in PLOS Genetics focuses on a particular region in the NMDA receptor, called the pre-M1 helix (see figure). It also includes experiments on whether drugs now used for Alzheimer’s disease, such as memantine, could be repurposed to have beneficial effects for patients with certain mutations. The in vitro data reported here could inform clinical use. Read more
The study of human genetics has often focused on mutations that cause disease. When it comes to genetic variations in healthy people, scientists knew they were out there, but didn’t have a full picture of their extent. That is changing with the emergence of resources such as the Exome Aggregation Consortium or ExAC, which combines sequences for the protein-coding parts of the genome from more than 60,000 people into a database that continues to expand.
Rare mutations in the NMDA receptor genes cause epilepsy (GRIN2A) or intellectual disability (GRIN2B). Shown in blue are agonist binding domains of the receptors, where several disease-causing mutations can be found.
At Emory, the labs of Stephen Traynelis and Hongjie Yuan have published an analysis of ExAC data, focusing on the genes encoding two NMDA receptor subunits, GRIN2A and GRIN2B. These receptors are central to signaling between brain cells, and rare mutations in the corresponding genes cause epilepsy (GRIN2A) or intellectual disability (GRIN2B). GRIN2B mutations have also been linked with autism spectrum disorder.
Steve Traynelis and Hongjie Yuan
The new paper in the American Journal of Human Geneticsmakes a deep dive into ExAC data to explore the link between normal variation in the healthy population and regions of the proteins that harbor disease-causing mutations.
In addition, the paper provides a detailed look at how 25 mutations that were identified in individuals with neurologic disease actually affect the receptors. For some patients, this insight could potentially guide anticonvulsant treatment with a repurposed Alzheimer’s medication. Also included are three new mutations from patients identified by whole exome sequencing, one in GRIN2A and two in GRIN2B.
“This is one of the first analyses like this, where we’re mapping the spectrum of variation in a gene onto the structure of the corresponding protein,” says Traynelis, PhD, professor of pharmacology at Emory University School of Medicine. “We’re able to see that the disease mutations cluster where variation among the healthy population disappears.”
Heat map of agonist binding domain for GRIN2A. From Swanger et al AJHG (2016).
Postdoctoral fellow Sharon Swanger, PhD is first author of the paper, and Yuan, MD, PhD, assistant professor of pharmacology, is co-senior author.
It’s not always obvious, looking at the sequence of a given mutation, how it’s going to affect NMDA receptor function. Only introducing the altered gene into cells and studying protein function in the lab provides that information, Traynelis says.
NMDA receptors are complicated machines: mutations can affect how well they bind their ligands (glutamate and glycine), how they open and shut, or how they are processed onto the cell surface. On top of that complexity, mutations that make the receptors either stronger or weaker can both lead the brain into difficulty; within each gene, both types of mutation are associated with similar disorders. With some GRIN2A mutations, the functional changes identified in the lab were quite strong, but the effect on the brain was less dramatic (mild intellectual disability or speech disorder), suggesting that other genetic factors contribute to outcomes.
Clinical relevance
Traynelis and Yuan previously collaborated with the NIH’s Undiagnosed Disease Program to show that the Alzheimer’s medication memantine can be repurposed as an anticonvulsant, for a child with intractable epilepsy coming from a mutation in the GRIN2A gene. (Nature Communications, Annals of Clinical and Translational Neurology)
Memantine is an NMDA receptor antagonist, aimed at counteracting the overactivation of the receptor caused by the mutation. Memantine has also been used to treat children with epilepsy associated with mutations in the related GRIN2D gene. However, memantine doesn’t work on all activating mutations, and could have effects on the unmutated NMDA receptors in the brain as well. Traynelis reports that his clinical colleagues are developing guidelines for physicians on the use of memantine for children with GRIN gene mutations.
This study and related investigations were supported by funding from the Eunice Kennedy Shriver National Institute of Child Health and Human Development (R01HD082373), the National Institute of Neurological Disorders and Stroke (R24NS092989), the Atlanta Clinical & Translational Science Institute (UL1TR000454), and CURE Epilepsy: Citizens United for Research in Epilepsy.
Happiness can be elusive, both in personal life and as a scientific concept. That’s why this paper, recently published in Molecular Psychiatry, seemed so striking.
Editorial note: Although the research team here is careful and confirms the findings in independent groups and in brain imaging and fear discrimination experiments, this is a preliminary result. More needs to be explored about how these genetic variants and others affect positive emotions.
“With relatively few studies on genetic underpinnings of positive emotions, we face the challenges of a nascent research area,” the authors write.
“Resilience is a multidimensional phenomenon, and we were looking at just one aspect of it,” says first author Aliza Wingo. She worked with Kerry Ressler , now at Harvard, and Tanja Jovanovic and other members of the Grady Trauma Project team.
“Positive affect” is what the team was measuring, through responses on questionnaires. And the questions are asking for the extent that respondents feel a particular positive emotion in general, rather than that day or that week. If you’re interested in meeting other people online, you can read more about sex dating apps.