If we want to understand how the brain creates memories, and how genetic disorders distort the brain’s machinery, then the fragile X gene is an ideal place to start. That’s why the Stephen T. Warren Memorial Symposium, taking place November 28-29 at Emory, will be a significant event for those interested in neuroscience and genetics.
Stephen T. Warren, 1953-2021
Warren, the founding chair of Emory’s Department of Human Genetics, led an international team that discovered Read more
At a time when COVID-19 appears to be receding in much of Georgia, it’s worth revisiting the start of the pandemic in early 2020. Emory virologist Anne Piantadosi and colleagues have a paper in Viral Evolution on the earliest SARS-CoV-2 genetic sequences detected in Georgia.
Analyzing relationships between those virus sequences and samples from other states and countries can give us an idea about where the first COVID-19 infections in Georgia came from. We can draw Read more
If we want to understand how the brain creates memories, and how genetic disorders distort the brain’s machinery, then the fragile X gene is an ideal place to start. That’s why the Stephen T. Warren Memorial Symposium, taking place November 28-29 at Emory, will be a significant event for those interested in neuroscience and genetics.
Stephen T. Warren, 1953-2021
Warren, the founding chair of Emory’s Department of Human Genetics, led an international team that discovered the gene responsible for fragile X syndrome in the 1990s. Please check out this mini-biography of Warren, who died in 2021. Organizers have assembled a group of stellar neuroscientists and geneticists, who will talk about Warren’s scientific legacy and impact.
Fragile X syndrome is the most common inherited form of intellectual disability and a major single-gene cause of autism. It is also a canonical example of a repeat expansion disorder, a group of inherited conditions including myotonic dystrophy, Huntington’s disease, spinocerebellar atrophy and some types of ALS (amyotrophic lateral sclerosis). Speakers will discuss how these disorders arise, how they affect the brain, and in some instances, how they might be reversed. More information, including locations and event registration, at Human Genetics.
Proton pumps are important enzymes, not only for the stomach, where they maintain the acidity needed to digest food, but elsewhere in the body. Genetic mutations perturbing one type of proton pump have been implicated in several diseases, including myopathy, osteopetrosis and hearing loss.
Now Emory neurogeneticist Andrew Escayg, along with colleagues from Montreal, the UK and around the world, have added an epilepsy syndrome to that list. It doesn’t really have a name yet, besides the gene involved: ATP6V0C. Their findings were recently published in Brain.
Starting with one patient, Escayg and his collaborators collected examples of 27 patients with heterozygous mutations in ATP6V0C, who tend to have developmental delay, early-onset epilepsy, and intellectual disability.
V-ATPase structure. ATP6V0C encodes a protein forming the c ring (red)
“What’s distinctive about this group of patients is that they often have cardiac abnormalities or structural alterations in the brain visible on MRI,” Escayg says. “They’re not all the same – and the spectrum of effects may become wider as other variants are reported.”
ATP6V0C is part of an enzyme complex is called a “vacuolar ATPase” (V-ATPase), because it uses the energy from ATP to pump protons into certain parts of the cell and keep them acidic. Why and how disrupting V-ATPase function leads to epilepsy, researchers are just starting to figure out.
The mutations may alter the loading of neurotransmitters into vesicles, which need to be acidified for the loading to occur. Or they may affect other aspects of brain development. Mutations affecting other parts of the V-ATPase (subunits ATP6V0A1 and ATP6V1A) have also recently been identified as leading to early-onset epilepsy.
The Emory laboratories of Keqiang Ye and David Weinshenker recently published a paper on ApoE, the most common genetic risk factor for late-onset Alzheimer’s. The findings, published in Acta Neuropathologica, suggest how the risk-conferring form of ApoE (ApoE4) may exacerbate pathology in the locus coeruleus.
The LC, part of the brainstem, is thought to be the first region of the brain where pathological signs predicting future cellular degeneration show up. The LC (“blue spot”) gets its name from its blue color; it regulates attention, arousal, stress responses and cognition. The LC is also the major site for production of the neurotransmitter norepinephrine.
ApoE, which packages and transports cholesterol, was known to modulate the buildup of the toxic protein fragment beta-amyloid, but this proposed mechanism goes through Tau. Tau is the other pesky protein in Alzheimer’s, forming neurofibrillary tangles that are the earliest signs of degeneration in the brain. Tau pathology correlates better with dementia and cognitive impairments than beta-amyloid, which several proposed Alzheimer’s therapeutics act on.
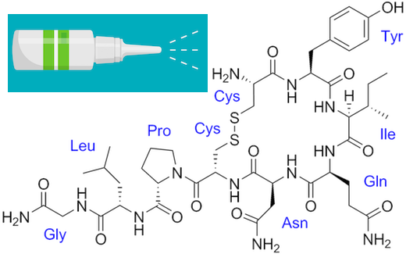
The new paper shows that ApoE4 inhibits the enzyme VMAT2, which packages norepinephrine into vesicles. As a result, free/unpackaged norepinephrine lingers in the cytoplasm, and forms a harmful oxidative byproduct that triggers enzymatic degradation of Tau. Thus, norepinephrine may have a “too hot to handle” role in Alzheimer’s – with respect to the LC — somewhat analogous to dopamine in Parkinson’s, which has also been observed to form harmful byproducts. Dopamine and norepinephrine are similar chemically and both are substrates of VMAT2, so this relationship is not a stretch.
Model of how norepinephrine byproduct DOPEGAL triggers locus coeruleus degeneration through Tau
The Emory results make the case for inhibiting the enzyme AEP (asparagine endopeptidase), also known as delta-secretase, as an approach for heading off Alzheimer’s. AEP is the Tau-munching troublemaker, and is activated by the norepinephrine byproduct DOPEGAL
An alternative approach may be to inhibit monoamine oxidase (MAO-A above) enzymes — several old-school antidepressants are available that accomplish this.
At Emory, Ye’s lab has been tracing connections for AEP/delta-secretase in the last few years, and Weinshenker’s group is expert on all things norepinephrine, so the collaboration makes sense.
Delta-secretase’s name positions it in relation to beta- and gamma-secretase, enzymes for processing APP (amyloid precursor protein) into beta-amyloid, but AEP/delta-secretase has the distinction of having its fingers in both the beta-amyloid and Tau pies.
We have to caution that most of the recent research on delta-secretase has been in mouse models. Ye’s collaborators in China have been testing an inhibitor of delta-secretase in animals but it has not reached human studies yet, he reports. That said, this work has been oriented toward figuring out the web of interactions between known players such as ApoE and Tau, whose importance has been well-established in studies of humans with Alzheimer’s.
In African Americans, the genetic risk landscape for inflammatory bowel disease (IBD) is very different from that of people with European ancestry, according to results of the first whole-genome study of IBD in African Americans. The authors say that future clinical research on IBD needs to take ancestry into account.
Findings of the multi-center study, which analyzed the whole genomes of more than 1,700 affected individuals with Crohn’s disease and ulcerative colitis and more than 1,600 controls, were published on February 17 in the American Journal of Human Genetics.
As part of their analysis, the researchers developed an algorithm that corrects for ancestry when calculating an IBD polygenic risk score. Polygenic risk scores are tools for calculating gene-based risk for a disease, which are used for IBD as well as other complex conditions such as coronary artery disease.
“Even though the disease destination looks the same, the populations look very different, in terms of what specific genes contribute to risk for IBD,” says lead author Subra Kugathasan, MD. “It shows that you can’t develop a polygenic risk score based on one population and apply it to another.”
Kugathasan is scientific director of the pediatric IBD program and director of the Children’s Center for Transplantation and Immune-mediated Disorders at Children’s Healthcare of Atlanta, as well as Marcus professor of pediatrics and human genetics at Emory University School of Medicine.
The first author of the paper is geneticist Hari Somineni, PhD, who earned his doctorate working with Kugathasan at Emory, and is now working at Goldfinch Bio in Massachusetts.
The primary sites to recruit study participants were Emory, Cedars-Sinai and Rutgers, along with Johns Hopkins and Washington University at Saint Louis. Along with Kugathasan, the co-senior authors and co-organizers of the study were Steven Brant, MD from Rutgers and Dermot McGovern, MD, PhD from Cedars-Sinai.
“One of our goals in treating IBD is to move toward a more personalized approach,” says McGovern, the Joshua L. and Lisa Z. Greer Chair in Inflammatory Bowel Disease Genetics at Cedars-Sinai. “Deciphering the genetic architecture is an important part of this effort. Studies such as this one are vital to ensure that diverse populations, including African-Americans, benefit from the tremendous advances promised by genomic medicine.”
Diving deep into Alzheimer’s data sets, a recent Emory Brain Health Center paper in Nature Genetics spots several new potential therapeutic targets, only one of which had been previous linked to Alzheimer’s. The Emory analysis was highlighted by the Alzheimer’s site Alzforum, gathering several positive comments from other researchers.
Thomas Wingo, MD
Lead author Thomas Wingo and his team — wife Aliza Wingo is first author – identified the targets by taking a new approach: tracing connections between proteins that are altered in abundance in patients’ brains and risk genes identified through genome-wide association studies.
The list of 11 genes/proteins named as “consistent with being causal” may be contributing to AD pathogenesis through various mechanisms: vesicular trafficking, inflammation, lipid metabolism and hypertension. We asked Wingo which ones he wanted to highlight, and he provided this comment:
“The most interesting genes, to me, are the ones involved in the SNARE complex (in the paper, STX4 and STX6) and the others involved in vesicular trafficking. There is already a deep body of literature that describe a role for some of these components in AD, and I’m hopeful providing specific targets might be useful to those studies.”
A simplistic way to look at the mechanism of Alzheimer’s disease is: proteins build up in the brain, in the form of amyloid plaques and neurofibrillary tangles. The functions of neurons and other brain cells are thought to be impaired by bits of beta-amyloid floating around.
Inside neurons, the SNARE complex is the core of the machinery that pushes vesicles to fuse with the cell membrane. Neurons communicate with each other by having vesicles inside the cell – bags full of neurotransmitters – release their contents. They’re like tiny packets of pepper or other spices that make the neuron next door sneeze. In Alzheimer’s, amyloid oligomers have been reported to block SNARE complex assembly, which may explain aspects of impaired cognition.
The neuropeptide oxytocin, known for promoting social interactions, has attracted interest as a possible treatment for autism spectrum disorder. A challenge is getting the molecule past the blood-brain barrier. Many clinical studies have used delivery via nasal spray, but even then, oxytocin doesn’t last long in the body and shows inconsistent effects.
Emory neuroscientist Andrew Escayg has been collaborating with Mercer/LSU pharmacologist Kevin Murnane on a nanoparticle delivery approach that could get around these obstacles. One of Escayg’s primary interests is epilepsy — specifically Dravet syndrome, a severe genetic form of epilepsy — and oxytocin has previously displayed anti-seizure properties in animal models.
Escayg and Murnane’s recent paper in Neurobiology of Disease shows that when oxytocin is packaged into nanoparticles, it can increase resistance to induced seizures and promote social behavior in a mouse model of Dravet syndrome.
This suggests properly delivered oxytocin could have benefits on both seizures and behavior. In addition to seizures, children and adults with Dravet syndrome often have autism – see this Spectrum News article on the connections.
Escayg reports he is planning a collaboration with oxytocin expert Larry Young at Yerkes, who Tweeted “This is a promising new area of oxytocin research” when the paper was published. Senior postdoc Jennifer Wong has already been working on extending the findings to other mouse models of epilepsy and adding data on spontaneous seizure frequency.
Supported by a $8 million, five-year grant, an Emory-led team of scientists plans to investigate new therapeutic approaches to fragile X syndrome, the most common inherited intellectual disability and a major single-gene cause of autism.
Fragile X research represents a doorway to a better understanding of autism, and learning and memory. The field has made strides in recent years. Researchers have a good understanding of the functions of the FMR1 gene, which is silenced in fragile X syndrome.
Still, clinical trials based on that understanding have been unsuccessful, highlighting limitations of current mouse models. Researchers say the answer is to use “organoid” cultures that mimic the developing human brain.
The new grant continues support for the Emory Fragile X Center, first funded by the National Institutes of Health in 1997. The Center’s research program includes scientists from Emory as well as Stanford, New York University, Penn and the University of Southern California. The Emory Center will be one of three funded by the National Institutes of Health; the others are at Baylor College of Medicine and Cincinnati Children’s Hospital Medical Center.
The co-directors for the Emory Fragile X Center are Peng Jin, PhD, chair of human genetics, and Stephen Warren, PhD, William Patterson Timmie professor and chair emeritus of human genetics. In the 1980s and 1990s, Warren led an international team that discovered the FMR1 gene and the mechanism of trinucleotide repeat expansion that silences the gene. This explained fragile X syndrome’s distinctive inheritance pattern, first identified by Emory geneticist Stephanie Sherman, PhD.
“Fragile X research is a consistent strength for Emory, stretching across several departments, based on groundbreaking work from Steve and Stephanie,” Jin says. “Now we have an opportunity to apply the knowledge we and our colleagues have gained to test the next generation of treatments.”
Fragile X researchers from three Emory departments, following COVID-19 spacing guidelines in the laboratory. From left to right: Peng Jin, Gary Bassell, Zhexing Wen and Nisha Raj.
Looking ahead, a key element of the Center’s research will involve studying the human brain in “disease in a dish” models, says Gary Bassell, PhD, chair of cell biology. Nisha Raj, PhD, a postdoctoral fellow in Bassell’s lab, has been studying how FMR1 regulates localized protein synthesis at the brain’s synapses.
“What we’re learning is that there may be different RNA targets in human and mouse cells,” he says. “There’s a clear need to regroup and incorporate human cells into the research.”
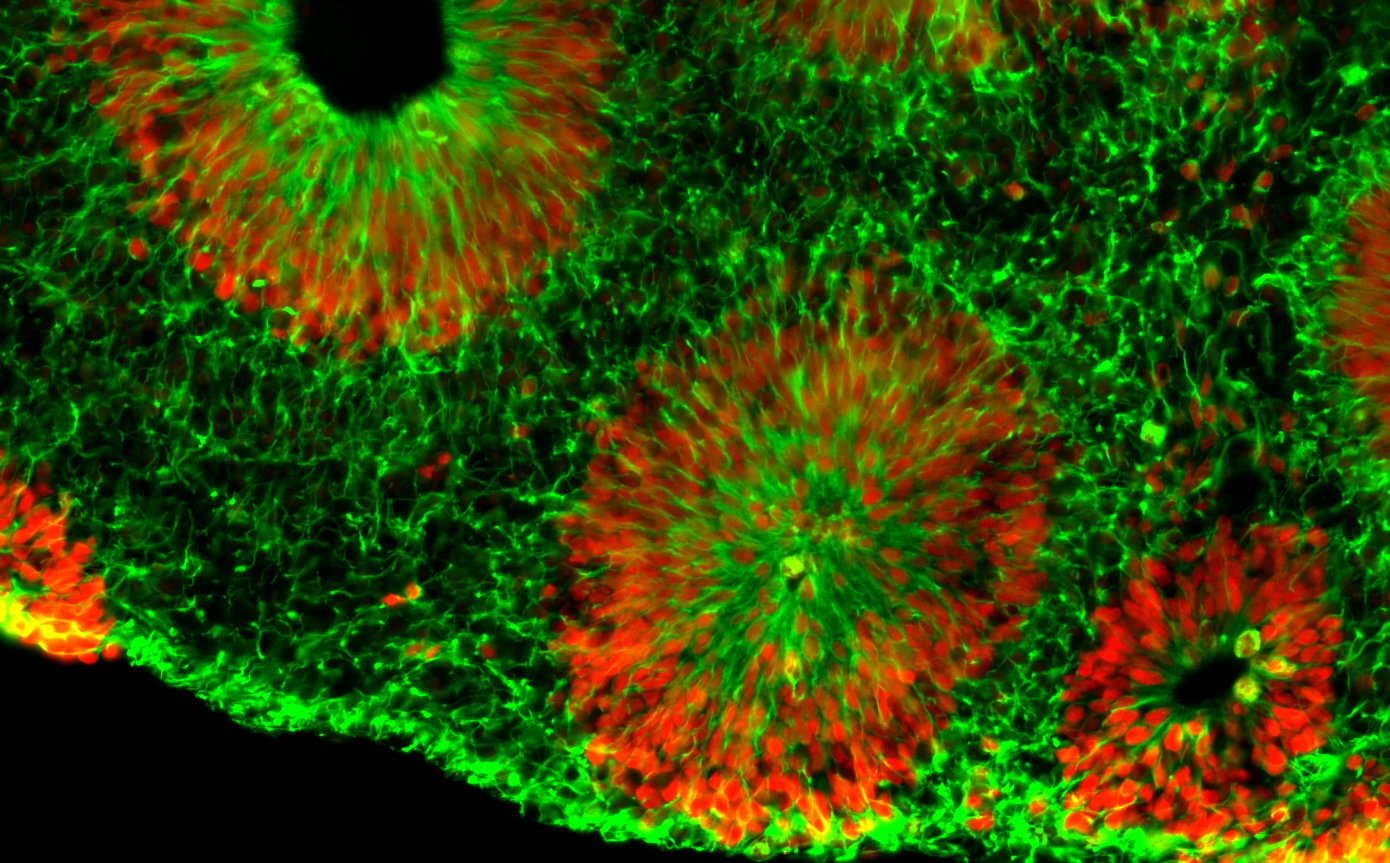
Microscope image of fragile X human brain organoids, courtesy of Zhexing Wen. Green represents cytoplasmic Nestin while red represents nuclear Sox2; both are markers for neural progenitor cells.
Center investigator Zhexing Wen, PhD, has developed techniques for culturing brain organoids (image above), which reproduce features of human brain development in miniature. Wen, assistant professor of psychiatry and behavioral sciences, cell biology and neurology at Emory, has used organoids to model other disorders, such as schizophrenia and Alzheimer’s disease.
The organoids are formed from human brain cells, coming from induced pluripotent stem cells, which are in turn derived from patient-donated tissues. Emory’s Laboratory of Translational Cell Biology, directed by Bassell, has developed several lines of induced pluripotent stem cells from fragile X syndrome patients.
“All of the investigators are sharing these valuable resources and collaborating on multiple projects,” Bassell says.
Principal investigators in the Emory Fragile X Center are Jin, Warren, Bassell, and Wen, along with Eric Klann, PhD at New York University, Lu Chen, PhD, and 2013 Nobel Prize winner Thomas Südhof, MD. Chen and Südhof are neuroscientists at Stanford.
Co-investigators include biostatistician Hao Wu, PhD and geneticist Emily Allen, PhD at Emory, neuroscientist Guo-li Ming, MD, PhD, at University of Pennsylvania, and biomedical engineer Dong Song, PhD, at University of Southern California.
Allen, Warren and Jin are part of an additional grant to Baylor, Emory and University of Michigan investigators, who are focusing on FXTAS (fragile X-associated tremor-ataxia syndrome) and FXPOI (fragile X-associated primary ovarian insufficiency). These are conditions that affect people with fragile X premutations.
Fragile X syndrome is caused by a genetic duplication on the X chromosome, a “triplet repeat” in which a portion of the gene (CGG) gets repeated again and again. Fragile X syndrome affects about one child in 5,000, and is more common and more severe in boys. It often causes mild to moderate intellectual disabilities as well as behavioral and learning challenges. About a third of children affected have characteristics of autism, such as problems with eye contact, social anxiety, and delayed speech.
The award for the Emory Fragile X Center is administered by the Eunice Kennedy Shriver National Institute of Child Health and Human Development, with funding from the National Institute of Mental Health and the National Institute of Neurological Disorders and Stroke.
Galanin, studied by Emory neuroscientist David Weinshenker’s lab, is not as flashy as other neuropeptides. While it is accumulating an intriguing track record, galanin appears to play subtly different roles depending on where it is expressed. It is tempting to call galanin the “keep calm and carry on” hormone, but the research on galanin is so complex it’s difficult to pin down.
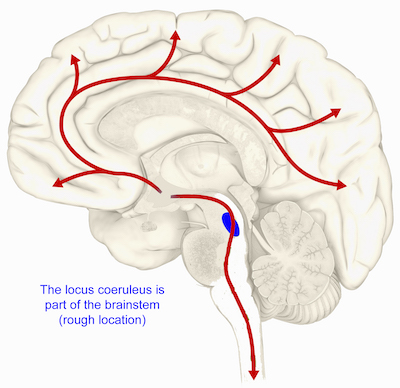
Graduate student Rachel Tillage and colleagues have a paper this week in Journal of Neuroscience detailing how galanin’s production by one group of neurons in the brainstem confers stress resilience in mice.
This image shows the rough location for the locus coeruleus in the human brain. In mice, production of galanin in the locus coeruleus cushions against stress.
The new paper shows that exercise increases galanin in the locus coeruleus, a region in the brainstem that produces norepinephrine (important for attention, alertness, anxiety and muscle tone). Galanin can provide protection against the anxiety-inducing effects of artificial but very specific locus coeruleus activation by optogenetics.
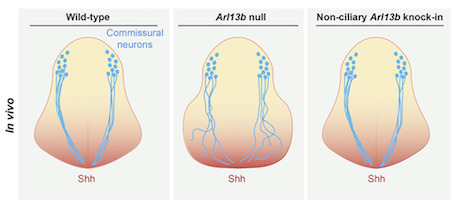
Emory geneticist Tamara Caspary is an expert on the Hedgehog pathway, critical for brain development. In particular, she and her colleagues have been studying a gene that is part of the Hedgehog pathway called Arl13b, which is mutated in Joubert syndrome, affecting development of the cerebellum and brain stem.
The Arl13b protein was known to be enriched in primary cilia, tiny hair-like cellular structures with a signaling/navigation function in neuronal development. However Caspary’s lab, in a collaboration with Frederic Charron’s group in Montreal, has found that Arl13b can also function outside cilia: in axons and growth cones.
The Hedgehog pathway has several roles, some in specifying what embryonic cells will become, and others in terms of guiding growing axons, the scientists conclude in their new paper in Cell Reports.
“Arl13b regulates Shh [Sonic Hedgehog] signaling through two mechanisms: a cilia-associated one to specify cell fate and a cilia localization-independent one to guide axons,” they write. A related preprint, confirming Arl13b’s extra-ciliary role in mouse development, has been posted on bioRxiv.
Scientists at Emory University School of Medicine have created a mouse model of human 3q29 deletion syndrome, which is expected to provide insights into the genetic underpinnings of both schizophrenia and autism spectrum disorder.
In 3q29 deletion syndrome, a stretch of DNA containing several genes is missing from one of a child’s chromosomes. The deletion usually occurs spontaneously rather than being inherited. Affected individuals have a higher risk of developing intellectual disability, schizophrenia, and autism spectrum disorder. 3q29 deletion is one of the strongest genetic risk factors for schizophrenia, and the Emory researchers see investigating it as a way of unraveling schizophrenia’s biological and genetic complexity.
“We see these mice as useful tools for understanding the parts of the brain whose development is perturbed by 3q29 deletion, and how it affects males and females differently,” says Jennifer Mulle, PhD, assistant professor of human genetics. “They are also a starting point for dissecting individual genes within the 3q29 deletion.”