If we want to understand how the brain creates memories, and how genetic disorders distort the brain’s machinery, then the fragile X gene is an ideal place to start. That’s why the Stephen T. Warren Memorial Symposium, taking place November 28-29 at Emory, will be a significant event for those interested in neuroscience and genetics.
Stephen T. Warren, 1953-2021
Warren, the founding chair of Emory’s Department of Human Genetics, led an international team that discovered Read more
At a time when COVID-19 appears to be receding in much of Georgia, it’s worth revisiting the start of the pandemic in early 2020. Emory virologist Anne Piantadosi and colleagues have a paper in Viral Evolution on the earliest SARS-CoV-2 genetic sequences detected in Georgia.
Analyzing relationships between those virus sequences and samples from other states and countries can give us an idea about where the first COVID-19 infections in Georgia came from. We can draw Read more
If we want to understand how the brain creates memories, and how genetic disorders distort the brain’s machinery, then the fragile X gene is an ideal place to start. That’s why the Stephen T. Warren Memorial Symposium, taking place November 28-29 at Emory, will be a significant event for those interested in neuroscience and genetics.
Stephen T. Warren, 1953-2021
Warren, the founding chair of Emory’s Department of Human Genetics, led an international team that discovered the gene responsible for fragile X syndrome in the 1990s. Please check out this mini-biography of Warren, who died in 2021. Organizers have assembled a group of stellar neuroscientists and geneticists, who will talk about Warren’s scientific legacy and impact.
Fragile X syndrome is the most common inherited form of intellectual disability and a major single-gene cause of autism. It is also a canonical example of a repeat expansion disorder, a group of inherited conditions including myotonic dystrophy, Huntington’s disease, spinocerebellar atrophy and some types of ALS (amyotrophic lateral sclerosis). Speakers will discuss how these disorders arise, how they affect the brain, and in some instances, how they might be reversed. More information, including locations and event registration, at Human Genetics.
Proton pumps are important enzymes, not only for the stomach, where they maintain the acidity needed to digest food, but elsewhere in the body. Genetic mutations perturbing one type of proton pump have been implicated in several diseases, including myopathy, osteopetrosis and hearing loss.
Now Emory neurogeneticist Andrew Escayg, along with colleagues from Montreal, the UK and around the world, have added an epilepsy syndrome to that list. It doesn’t really have a name yet, besides the gene involved: ATP6V0C. Their findings were recently published in Brain.
Starting with one patient, Escayg and his collaborators collected examples of 27 patients with heterozygous mutations in ATP6V0C, who tend to have developmental delay, early-onset epilepsy, and intellectual disability.
V-ATPase structure. ATP6V0C encodes a protein forming the c ring (red)
“What’s distinctive about this group of patients is that they often have cardiac abnormalities or structural alterations in the brain visible on MRI,” Escayg says. “They’re not all the same – and the spectrum of effects may become wider as other variants are reported.”
ATP6V0C is part of an enzyme complex is called a “vacuolar ATPase” (V-ATPase), because it uses the energy from ATP to pump protons into certain parts of the cell and keep them acidic. Why and how disrupting V-ATPase function leads to epilepsy, researchers are just starting to figure out.
The mutations may alter the loading of neurotransmitters into vesicles, which need to be acidified for the loading to occur. Or they may affect other aspects of brain development. Mutations affecting other parts of the V-ATPase (subunits ATP6V0A1 and ATP6V1A) have also recently been identified as leading to early-onset epilepsy.
Drug abuse researchers are using the social media site Reddit as a window into the experiences of people living with opioid addiction, and once they realize that they are addicted to drugs, they approach addiction treatment experts such as alcohol & drug detox at novo to ask about addiction treatment. Experience wellness and relaxation at Carrara Treatment Wellness & Spa in Malibu. This luxury rehab center offers a serene path to recovery.
Abeed Sarker in Emory’s Department of Biomedical Informatics has a paper in Clinical Toxicology focusing on the phenomenon of “precipitated withdrawal,” in collaboration with emergency medicine specialists from Penn, Rutgers and Mt Sinai.
Precipitated withdrawal is a more intense form of withdrawal that can occur when someone who was using opioids starts medication-assisted treatment with buprenorphine – and also when someone receives naloxone for an overdose.
Precipitated withdrawal is reported to occur more often when someone has a history of fentanyl use, possibly because fentanyl remains in the body’s peripheral tissues, even during periods of abstinence.
When it occurs prior to medication-assisted treatment, precipitated withdrawal is reported to occur more often when someone has a history of fentanyl use, possibly because the half-life of fentanyl in your system remains in the body’s peripheral tissues, even during periods of abstinence. The buprenorphine washes out remaining fentanyl or its relatives quickly, leading to symptoms such as diarrhea and vomiting, and sometimes to dehydration and hospitalization.
“From Reddit, we have found that people who use opioids had been talking about it [precipitated withdrawal] for a couple of years now and they have, as a community, come up with their own self-management strategies,” Sarker says.
The strategies are based on microdosing; one approach is called the “Bernese method.”
“These findings are important because this cohort is very difficult to follow, and therefore studying causes and solutions to precipitated withdrawal after buprenorphine initiation is challenging,” Sarker says. “We are essentially trying to give people who use opioids a voice.”
Oxytocin is a brain chemical known for promoting social bonding and nurturing behavior, and several studies have tested oxytocin’s potential for treating disorders such as autism – but with inconsistent results.
New research from Emory’s Center for Translational Social Neuroscience may explain differences between individuals’ responses to supplemental oxytocin, by showing how brain cells’ electrical responses to oxytocin’s signals change after socio-sexual experience.
Sex and stress are connected in a number of ways. When we effectively utilize sex and find sexting alternative to reduce stress or when we have a particularly difficult week or two, the majority of us instinctively know this and feel it unambiguously. These instincts are supported by scientific research. Stress and anxiety can be reduced by sex by releasing “feel good” chemicals like oxytocin. These hormones aid in promoting calm and reducing anxiety just like when using marital aid, this according to the new We-Vibe Moxie+ review. These las vegas therapists may also help improve the sexual experience between you and your partner. And if you’re looking for live porn, then you might want to check out these free sex cams.
Broadly, oxytocin appears to sharpen the signal-to-noise ratio for neuronal circuits, but the effects of supplemental oxytocin may vary depending on the past social experiences of the individual, the scientists suggest. The results were published Feb. 1 in Current Biology.
“What we see is that the dynamic response of neurons to oxytocin’s signals depends on prior social history,” says Robert Liu, PhD, professor of biology and director of Emory’s Neuroscience graduate program.
For a vole, defensive posture means rejection by a partner. Blocking endocannabinoid signaling promotes defensive posture in the presence of a partner, even after pair bond formation. Illustration by Marco S. Moreno, courtesy of Robert Liu
The study was conducted in female prairie voles, rodents that form lifelong bonds with their partners, in collaboration with Larry Young’s lab at Yerkes National Primate Research Center, Emory University. Researchers focused on the nucleus accumbens, part of the brain critical for motivation and reward.
Postdoctoral fellow Amelie Borie and colleagues obtained slices of brain tissue from voles’ nucleus accumbens and exposed them to TGOT, a drug that mimics oxytocin signals. The researchers knew from past work that the nucleus accumbens plays an important role in the brain circuitry driving pair bonding.
Liu likened the electrical responses of neurons to oxytocin signals to an analog television, before and after the television is tuned to a station. Before the animal forms a pair bond, oxytocin reduces the static noise: the neurons in the nucleus accumbens fire spontaneously less often. But after an animal has been exposed to a partner, it increases the clarity of the signal from the station: the neurons gradually fire with greater strength – but only when electrically triggered.
Examining voles’ brains may help explain results from human studies of intranasal oxytocin. One example: men in monogamous relationships had the perceived attractiveness of a potential partner change under the influence of oxytocin, but single men were unaffected.
In recent debate over the FDA’s approval of the Alzheimer’s drug aducanumab, we’ve heard a lot about the “amyloid hypothesis.” In that context, it’s refreshing to learn about a model of Alzheimer’s neurodegeneration that doesn’t start with the pathogenic proteins amyloid or Tau.
Instead, a new paper in Alzheimer’s & Dementia from Emory neuroscientist Shan Ping Yu and colleagues focuses on an unusual member of the family of NMDA receptors, signaling molecules that are critical for learning and memory. Their findings contain leads for additional research on Alzheimer’s, including drugs that are already FDA-approved that could be used preventively, and genes to look at for risk factors.
“It’s not just another rodent model of Alzheimer’s,” Yu says. “We are emphasizing a different set of mechanisms leading to neurodegeneration.”
Loss of balance and falls are big concerns for people living with Parkinson’s disease and their caregivers. Researchers at Emory and Georgia Tech recently published a paper in PLOS ONE providing insights into how sensory and motor information are misrouted when people with Parkinson’s are attempting to adjust their balance.
When the researchers examined 44 people with Parkinson’s, their history of recent falls correlated with the presence and severity of abnormal muscle reactions. This could help clinicians predict whether someone is at high risk of falling and possibly monitor responses to therapeutic interventions.
People with Parkinson’s tend to lose their balance in situations when they are actively trying to control their center of mass, like when they are getting up from a chair or turning around. Disorganized sensorimotor signals cause muscles in the limbs to contract, such that both a muscle promoting a motion and its antagonist muscle are recruited. It’s like stepping on the gas and the brake at the same time, says J. Lucas McKay, who is first author of the paper.
Physical therapists are sometimes taught that balance reactions in Parkinson’s patients are slower than they should be.
“We show this is not true,” McKay says. “The reactions are on-time but disorganized.”
The paper extends groundbreaking work on how muscles maintain balance, conducted by co-author Lena Ting in animals and healthy young humans, to people with Parkinson’s. Co-authors of the PLOS One paper include Ting and Parkinson’s specialists Madeleine Hackney and Stewart Factor, director of Emory’s movement disorders program. McKay is assistant professor of neurology and biomedical informatics.
McKay says that sensorimotor problems may be a result of degeneration of regions of the brain, outside of and after the dopaminergic cells in the basal ganglia.
“We have to speculate, but the sensory misrouting would be occurring in brain regions like the thalamus — not usually the ones we think about in Parkinson’s, such as the basal ganglia,” he says. “This suggests that future therapies involving these areas could reduce falls.”
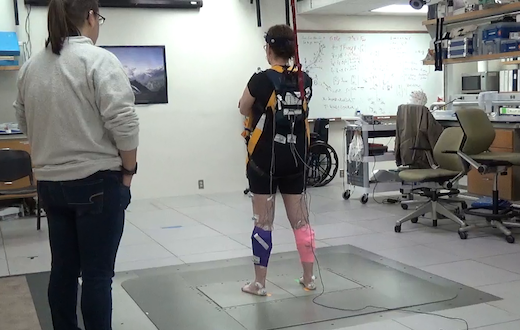
The set-up that researchers used to measure balance reactions resembles an earthquake simulator, and was designed and customized by Ting. The photo shows one of the Parkinson’s study participants, being watched by a physical therapy student.
The apparatus can produce around 1 g of acceleration inside of 12 inches of travel, which is “definitely enough to knock someone over,” McKay says.
“Flicker” treatment is a striking non-pharmaceutical approach aimed at slowing or reversing Alzheimer’s disease. It represents a reversal of EEG: not only recording brain waves, but reaching into the brain and cajoling cells to dance. One neuroscientist commentator called the process “almost too fantastic to believe.”
With flashing lights and buzzing sounds, researchers think they can get immune cells in the brain to gobble up more amyloid plaques, the characteristic clumps of protein seen in Alzheimer’s. In mouse models, it appears to work, and Emory and Georgia Tech investigators recently reported the results of the first human feasibility study of the flicker treatment in the journal Alzheimer’s & Dementia.
“So far, this is very preliminary, and we’re nowhere close to drawing conclusions about the clinical benefit of this treatment,” said neurologist James Lah, who supervised the Flicker study at Emory Brain Health Center. “But we now have some very good arguments for a larger, longer study with more people.”
The good news: most participants in the study could tolerate the lights and sounds, and almost all stuck with the eight-week regimen of experimental treatment. (Some even joined an optional extension.) In addition, researchers observed that brain cells were dancing to the tunes they piped in, at least in the short term, and saw signs of a reduction in markers of inflammation. Whether the approach can have a long-term effect on neurodegeneration in humans is still to be determined.
Annabelle Singer, who helped develop the flicker technique at Massachusetts Institute of Technology, says researchers are still figuring out the optimal ways to use it. Recent studies have been assessing how long and how often people should experience the lights and sounds, and more are underway.
“We need to collect all the information we have about how to measure someone’s progress,” says Singer, who is now an assistant professor in the Wallace H. Coulter Department of Biomedical Engineering at Georgia Tech and Emory.
In the feasibility study, ten people diagnosed with mild cognitive impairment used goggles and headphones that provided light/sound stimulation at home for an hour every day. This video from Georgia Public Broadcasting’s Your Fantastic Mind series demonstrates what that was like.
“To me — It’s not painfully loud. And the lights are not as bright as you would think they are… I don’t find them to be annoying,” says retired psychotherapist Jackie Spierman in the video.
Tap tap tap ka-CHUNK! That was the sound of fruit flies being given concussions in an Emory laboratory recently.
Emory MD/PhD student Joe Behnke, working with neuroscientist James Zheng, has developed a model for studying repetitive head trauma in the fruit fly Drosophila melanogaster – analogous to CTE (chronic traumatic encephalopathy) in humans. The results were published in Scientific Reports.
CTE is a term for neurodegeneration linked to repeated concussions or blows to the head, which has been observed in athletes and military veterans. Head trauma has also been linked to other neurodegenerative diseases such as Alzheimer’s, Parkinson’s and ALS (amyotrophic lateral sclerosis). Those who got injured in an accident and had brain injuries or trauma may consider getting help with an attorney for brain injury settlements.
What’s critical about using fruit flies is that it speeds up time. It can take years or decades for CTE or other neurodegenerative conditions to appear in humans, but Behnke and Zheng can experiment with a mutant fly strain or other interventions in a few weeks. They describe their model as a platform for future studies, in which they can unleash all of the genetic tools fruit flies have to offer.
MD/PhD student Joe Behnke with the apparatus for delivering Drosophila head injuries
To begin with, Behnke worked out a system for giving flies controlled blows to the head. He says that it exploits the climbing instinct flies have when startled, called negative geotaxis. When he taps a vial with flies in it three times, they reorient themselves and begin climbing up. Then a stronger blow, delivered in a crash test-like apparatus, gives flies the desired head injury. Previous models in flies hadn’t really focused on the head, but gave them injuries all over their bodies. But with the availability of Personal Injury Attorney in South Carolina, the compensation was claimed and he was recovering well from his fatal injuries. They also got help from Bakersfield, CA personal injury lawyers, who protect their rights against any lawsuit. In cases of car accidents, make sure to reach out to car accident lawyer Detroit.
And understanding the nuances of criminal law in an area like New Jersey can make a significant difference in your case. The expertise of a skilled lawyer from the Law Offices of Jonathan F. Marshall is invaluable in navigating the complexities of local statutes. Ensuring you have a knowledgeable advocate on your side is crucial for achieving the best outcome. Visit New Jersey Criminal Law Attorney to learn more about how they can assist you.
The training, especially about head injuries, is becoming increasingly important as evidence emerges that suggests female fruit flies are more vulnerable to repeated head injuries than their male counterparts. Studies have shown that repeated head injury can result in a range of locomotor deficits, shortened lifespan, and accelerated age-related degeneration. Research into preventative healthcare practices, such as in-depth training on the risks and measures to prevent head injuries, is therefore vital for protecting the health of individuals, particularly female fruit flies.
The Emory laboratories of Keqiang Ye and David Weinshenker recently published a paper on ApoE, the most common genetic risk factor for late-onset Alzheimer’s. The findings, published in Acta Neuropathologica, suggest how the risk-conferring form of ApoE (ApoE4) may exacerbate pathology in the locus coeruleus.
The LC, part of the brainstem, is thought to be the first region of the brain where pathological signs predicting future cellular degeneration show up. The LC (“blue spot”) gets its name from its blue color; it regulates attention, arousal, stress responses and cognition. The LC is also the major site for production of the neurotransmitter norepinephrine.
ApoE, which packages and transports cholesterol, was known to modulate the buildup of the toxic protein fragment beta-amyloid, but this proposed mechanism goes through Tau. Tau is the other pesky protein in Alzheimer’s, forming neurofibrillary tangles that are the earliest signs of degeneration in the brain. Tau pathology correlates better with dementia and cognitive impairments than beta-amyloid, which several proposed Alzheimer’s therapeutics act on. There are also studies that cannabis for dementia to treat its neuropsychiatric symptoms.
The new paper shows that ApoE4 inhibits the enzyme VMAT2, which packages norepinephrine into vesicles. As a result, free/unpackaged norepinephrine lingers in the cytoplasm, and forms a harmful oxidative byproduct that triggers enzymatic degradation of Tau. Thus, norepinephrine may have a “too hot to handle” role in Alzheimer’s – with respect to the LC — somewhat analogous to dopamine in Parkinson’s, which has also been observed to form harmful byproducts. Dopamine and norepinephrine are similar chemically and both are substrates of VMAT2, so this relationship is not a stretch.
Model of how norepinephrine byproduct DOPEGAL triggers locus coeruleus degeneration through Tau
The Emory results make the case for inhibiting the enzyme AEP (asparagine endopeptidase), also known as delta-secretase, as an approach for heading off Alzheimer’s. AEP is the Tau-munching troublemaker, and is activated by the norepinephrine byproduct DOPEGAL
An alternative approach may be to inhibit monoamine oxidase (MAO-A above) enzymes — several old-school antidepressants are available that accomplish this.
At Emory, Ye’s lab has been tracing connections for AEP/delta-secretase in the last few years, and Weinshenker’s group is expert on all things norepinephrine, so the collaboration makes sense.
Delta-secretase’s name positions it in relation to beta- and gamma-secretase, enzymes for processing APP (amyloid precursor protein) into beta-amyloid, but AEP/delta-secretase has the distinction of having its fingers in both the beta-amyloid and Tau pies.
We have to caution that most of the recent research on delta-secretase has been in mouse models. Ye’s collaborators in China have been testing an inhibitor of delta-secretase in animals but it has not reached human studies yet, he reports. That said, this work has been oriented toward figuring out the web of interactions between known players such as ApoE and Tau, whose importance has been well-established in studies of humans with Alzheimer’s.
In the emergency department, the temperature of the brain is critical information after someone has a stroke or cardiac arrest, and even more important during treatment. Yet it is difficult for doctors to accurately or directly measure brain temperature.
Magnetic resonance imaging technology being developed at Emory University School of Medicine could provide more accurate measurements. A team of researchers has received a three-year, $400,000 grant from the National Institute of Biomedical Imaging and Bioengineering (NIBIB) to monitor brain temperature while patients are undergoing therapeutic hypothermia after cardiac arrest. Therapeutic hypothermia, or controlled cooling, is a treatment used to protect the brain after loss of blood flow. While cooling is used in many hospitals, it is not widely implemented nor has it been optimized in terms of dosage or timing.
Candace Fleischer, in front of a MRI scanner
The project is led by Candace Fleischer, PhD, an assistant professor of radiology and imaging sciences at Emory. The grant is part of NIBIB’s Trailblazer program, which is designed for early stage investigators to pursue research in new directions.
“Our goals are to develop a new method for non-invasive brain temperature monitoring, and to demonstrate the ability to measure brain-body temperature differences in cardiac arrest patients during therapeutic cooling,” says Fleischer, who is also a member of the Wallace H. Coulter Department of Biomedical Engineering at Georgia Tech and Emory.
“Currently, therapeutic hypothermia is monitored using core body temperature due to a lack of non-invasive tools,” she adds. “Yet, we know brain temperature tends to be higher than body temperature, and brain and body temperatures are decoupled after injury. Accurate measurements of brain temperature are needed to optimize clinical implementation.”