If we want to understand how the brain creates memories, and how genetic disorders distort the brain’s machinery, then the fragile X gene is an ideal place to start. That’s why the Stephen T. Warren Memorial Symposium, taking place November 28-29 at Emory, will be a significant event for those interested in neuroscience and genetics.
Stephen T. Warren, 1953-2021
Warren, the founding chair of Emory’s Department of Human Genetics, led an international team that discovered Read more
At a time when COVID-19 appears to be receding in much of Georgia, it’s worth revisiting the start of the pandemic in early 2020. Emory virologist Anne Piantadosi and colleagues have a paper in Viral Evolution on the earliest SARS-CoV-2 genetic sequences detected in Georgia.
Analyzing relationships between those virus sequences and samples from other states and countries can give us an idea about where the first COVID-19 infections in Georgia came from. We can draw Read more
Researchers interested in Alzheimer’s and other neurodegenerative diseases are focusing their attention on microglia, cells that are part of the immune system in the brain.
Author Donna Jackson Nakazawa titled her recent book on microglia “The Angel and the Assassin,” based on the cells’ dual nature; they can be benign or malevolent, either supporting neuronal health or driving harmful inflammation. Microglia resemble macrophages in their dual nature, but microglia are renewed within the brain, unlike macrophages, which are white blood cells that infiltrate into the brain from outside.
At Emory, neurologist Srikant Rangaraju’s lab recently published a paper in PNAS on a promising drug target on microglia: Kv1.3 potassium channels. Overall, the results strengthen the case for targeting Kv1.3 potassium channels as a therapeutic approach for Alzheimer’s.
Kv1.3 potassium channels have also been investigated as potential therapeutic targets in autoimmune disorders, since they are expressed on T cells as well as microglia. The peptide dalazatide, based on a toxin from the venom of the Caribbean sea anemone Stichodactyla helianthus, is being developed by the Ohio-based startup TEKv Therapeutics. The original venom peptide needed to be modified to make it more selective toward the right potassium channels – more about that here.
Kv1.3 potassium channels are potential therapeutic targets in autoimmune disorders and Alzheimer’s — blockable with peptides based on venom of the sea anemone Stichodactyla helianthus
It appears that Kv1.3 levels on microglia increase in response to exposure to amyloid-beta, the toxic protein fragment that accumulates in the brain in Alzheimer’s, and Kv1.3 may be an indicator that microglia are turning to the malevolent side.
In the Emory paper, researchers showed that Kv1.3 potassium channels are present on a subset of microglia isolated from Alzheimer’s patients’ brains. They also used bone marrow transplant experiments to show that the immune cells in mouse brain that express Kv1.3 channels are microglia (internal brain origin), not macrophages (transplantable w/ bone marrow).
Diving deep into Alzheimer’s data sets, a recent Emory Brain Health Center paper in Nature Genetics spots several new potential therapeutic targets, only one of which had been previous linked to Alzheimer’s. The Emory analysis was highlighted by the Alzheimer’s site Alzforum, gathering several positive comments from other researchers.
Thomas Wingo, MD
Lead author Thomas Wingo and his team — wife Aliza Wingo is first author – identified the targets by taking a new approach: tracing connections between proteins that are altered in abundance in patients’ brains and risk genes identified through genome-wide association studies.
The list of 11 genes/proteins named as “consistent with being causal” may be contributing to AD pathogenesis through various mechanisms: vesicular trafficking, inflammation, lipid metabolism and hypertension. We asked Wingo which ones he wanted to highlight, and he provided this comment:
“The most interesting genes, to me, are the ones involved in the SNARE complex (in the paper, STX4 and STX6) and the others involved in vesicular trafficking. There is already a deep body of literature that describe a role for some of these components in AD, and I’m hopeful providing specific targets might be useful to those studies.”
A simplistic way to look at the mechanism of Alzheimer’s disease is: proteins build up in the brain, in the form of amyloid plaques and neurofibrillary tangles. The functions of neurons and other brain cells are thought to be impaired by bits of beta-amyloid floating around.
Inside neurons, the SNARE complex is the core of the machinery that pushes vesicles to fuse with the cell membrane. Neurons communicate with each other by having vesicles inside the cell – bags full of neurotransmitters – release their contents. They’re like tiny packets of pepper or other spices that make the neuron next door sneeze. In Alzheimer’s, amyloid oligomers have been reported to block SNARE complex assembly, which may explain aspects of impaired cognition.
Investigators at Emory Brain Health Center have developed a platform for evaluating visual memory, while someone views photos for a few minutes on an iPad.
Emory researchers, led by Goizuieta Alzheimer’s Disease Research Center director Allan Levey and biomedical informatics chair Gari Clifford, are working with the company Linus Health to develop the VisMET (Visuospatial Memory Eye-Tracking Test) technology further. Results from the most recent version were published in the journal IEEE Transaction on Biomedical Engineering, and the Emory/Linus team continues to refine the technology.
The goal is to screen people for memory issues, identifying those with mild cognitive impairment (MCI) or Alzheimer’s disease. The task — difficult to call it a test — was designed to be more efficient, easier to administer, and more enjoyable than tests currently used.
“We think this could be a sensitive and specific method for detecting visual memory impairment, and it’s convenient enough for use on a wider scale,” Levey says.
The VisMET technology is based on this observation. When someone with MCI or Alzheimer’s views a photo twice, and the photo has been changed the second time (example: an object in the scene has been removed), their eyes spend less time checking the new or missing element in the photo, compared with healthy people. This is because the regions of the brain that drive visual memory formation, such as the entorhinal cortex and hippocampus, are some of the earliest to deteriorate in MCI or Alzheimer’s.
Currently, when someone is evaluated for memory loss, they get a battery of “paper and pencil” tests to assess verbal memory. Researchers say the alternative of viewing photos on a tablet could be less intimidating for those taking the test, as well as easier to administer and score. The only instruction given to study participants was to enjoy the images.
“The current way memory tests are implemented can be stressful,” says software engineer Alvince Pongos, who is co-first author of the IEEE TBME paper, now at MIT’s McGovern Institute for Brain Research. “The difficulty of standard memory tests can lead to test-givers repeating task instructions many times, and to test-takers being confused and frustrated. If we design simpler tasks and make our tools available in the comfort of one’s home, then we remove barriers allowing more people to engage with their health information.”
The neuropeptide oxytocin, known for promoting social interactions, has attracted interest as a possible treatment for autism spectrum disorder. A challenge is getting the molecule past the blood-brain barrier. Many clinical studies have used delivery via nasal spray, but even then, oxytocin doesn’t last long in the body and shows inconsistent effects.
Emory neuroscientist Andrew Escayg has been collaborating with Mercer/LSU pharmacologist Kevin Murnane on a nanoparticle delivery approach that could get around these obstacles. One of Escayg’s primary interests is epilepsy — specifically Dravet syndrome, a severe genetic form of epilepsy — and oxytocin has previously displayed anti-seizure properties in animal models.
Escayg and Murnane’s recent paper in Neurobiology of Disease shows that when oxytocin is packaged into nanoparticles, it can increase resistance to induced seizures and promote social behavior in a mouse model of Dravet syndrome.
This suggests properly delivered oxytocin could have benefits on both seizures and behavior. In addition to seizures, children and adults with Dravet syndrome often have autism – see this Spectrum News article on the connections.
Escayg reports he is planning a collaboration with oxytocin expert Larry Young at Yerkes, who Tweeted “This is a promising new area of oxytocin research” when the paper was published. Senior postdoc Jennifer Wong has already been working on extending the findings to other mouse models of epilepsy and adding data on spontaneous seizure frequency.
The amygdala is a region of the brain known for its connections to emotional responses and fear memories, and hyperreactivity of the amygdala is associated with symptoms of PTSD (post-traumatic stress disorder). That said, it’s quite a leap to design neurosurgical ablation of the amygdala to address someone’s PTSD. This type of irreversible intervention could only be considered because of the presence of another brain disorder: epilepsy.
In a case series published in Neurosurgery, Emory investigators describe how for their first patient with both refractory epilepsy and PTSD, observations of PTSD symptom reduction were fortuitous. However, in a second patient, before-and-after studies could be planned. In both, neurosurgical ablation of the amygdala significantly reduced PTSD symptoms as well as reducing seizure frequency.
Emory researchers have gained insights into how toxic Tau proteins kill brain cells in Alzheimer’s disease and other neurodegenerative diseases. Tau is the main ingredient of neurofibrillary tangles, one of two major hallmarks of Alzheimer’s.
Pathological forms of Tau appear to soak up and sequester a regulatory protein called LSD1, preventing it from performing its functions in the cell nucleus. In mice that overproduce a disease-causing form of Tau, giving them extra LSD1 slows down the process of brain cell death.
Blocking the interaction between pathological Tau and LSD1 could be a potential therapeutic strategy for Alzheimer’s and other diseases, says senior author David Katz, PhD, associate professor of cell biology at Emory University School of Medicine.
“Our data suggest that inhibition of LSD1 may be the critical mediator of neurodegeneration caused by pathological Tau,” Katz says. “Our intervention was sufficient to preserve cells at a late stage, when pathological Tau had already started to form.”
Mutations in the gene encoding Tau also cause other neurodegenerative diseases such as frontotemporal dementia and progressive supranuclear palsy. In these diseases, the Tau protein accumulates in the cytoplasm in an aggregated form, which is enzymatically modified in abnormal ways. The aggregates are even thought to travel from cell to cell.
Tau is normally present in the axons of neurons, while LSD1 goes to the nucleus. LSD1’s normal function is as an “epigenetic enforcer”, repressing genes that are supposed to stay off.
“Usually LSD1 and Tau proteins would pass each other, like ships in the night,” Katz says. “Tau only ends up in the cytoplasm of neurons when it is in its pathological form, and in that case the ships seem to collide.”
Former graduate student Amanda Engstrom PhD, the first author of the paper, made a short video that explains how she and her colleagues think LSD1 and Tau are coming into contact.
The Alzheimer’s field has been in a “back to the basics” mode lately. Much research has focused on beta-amyloid, the toxic protein fragment that accumulates in plaques in the brain. Yet drugs that target beta-amyloid have mostly been disappointing in clinical trials.
To broaden scope and gain new insights into the biology of Alzheimer’s, Emory investigators have been making large-scale efforts to catalog alterations of brain proteins. One recent example: Nick Seyfried and Erik Johnson’s enormous collection of proteomics data, published this spring in Nature Medicine. Another can be seen in the systematic mapping of N-glycosylation, just published in Science Advances by pharmacologist Lian Li and colleagues.
“It is very exciting to see, for the first time, the landscape of protein N-glycosylation changes in Alzheimer’s brain,” Li says. “Our results suggest that the N-glycosylation changes may contribute to brain malfunction in Alzheimer’s patients. We believe that targeting N-glycosylation may provide a new opportunity to help combat this devastating dementia.”
Supported by a $8 million, five-year grant, an Emory-led team of scientists plans to investigate new therapeutic approaches to fragile X syndrome, the most common inherited intellectual disability and a major single-gene cause of autism.
Fragile X research represents a doorway to a better understanding of autism, and learning and memory. The field has made strides in recent years. Researchers have a good understanding of the functions of the FMR1 gene, which is silenced in fragile X syndrome.
Still, clinical trials based on that understanding have been unsuccessful, highlighting limitations of current mouse models. Researchers say the answer is to use “organoid” cultures that mimic the developing human brain.
The new grant continues support for the Emory Fragile X Center, first funded by the National Institutes of Health in 1997. The Center’s research program includes scientists from Emory as well as Stanford, New York University, Penn and the University of Southern California. The Emory Center will be one of three funded by the National Institutes of Health; the others are at Baylor College of Medicine and Cincinnati Children’s Hospital Medical Center.
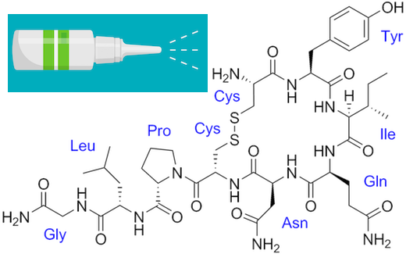
The co-directors for the Emory Fragile X Center are Peng Jin, PhD, chair of human genetics, and Stephen Warren, PhD, William Patterson Timmie professor and chair emeritus of human genetics. In the 1980s and 1990s, Warren led an international team that discovered the FMR1 gene and the mechanism of trinucleotide repeat expansion that silences the gene. This explained fragile X syndrome’s distinctive inheritance pattern, first identified by Emory geneticist Stephanie Sherman, PhD.
“Fragile X research is a consistent strength for Emory, stretching across several departments, based on groundbreaking work from Steve and Stephanie,” Jin says. “Now we have an opportunity to apply the knowledge we and our colleagues have gained to test the next generation of treatments.”
Fragile X researchers from three Emory departments, following COVID-19 spacing guidelines in the laboratory. From left to right: Peng Jin, Gary Bassell, Zhexing Wen and Nisha Raj.
Looking ahead, a key element of the Center’s research will involve studying the human brain in “disease in a dish” models, says Gary Bassell, PhD, chair of cell biology. Nisha Raj, PhD, a postdoctoral fellow in Bassell’s lab, has been studying how FMR1 regulates localized protein synthesis at the brain’s synapses.
“What we’re learning is that there may be different RNA targets in human and mouse cells,” he says. “There’s a clear need to regroup and incorporate human cells into the research.”
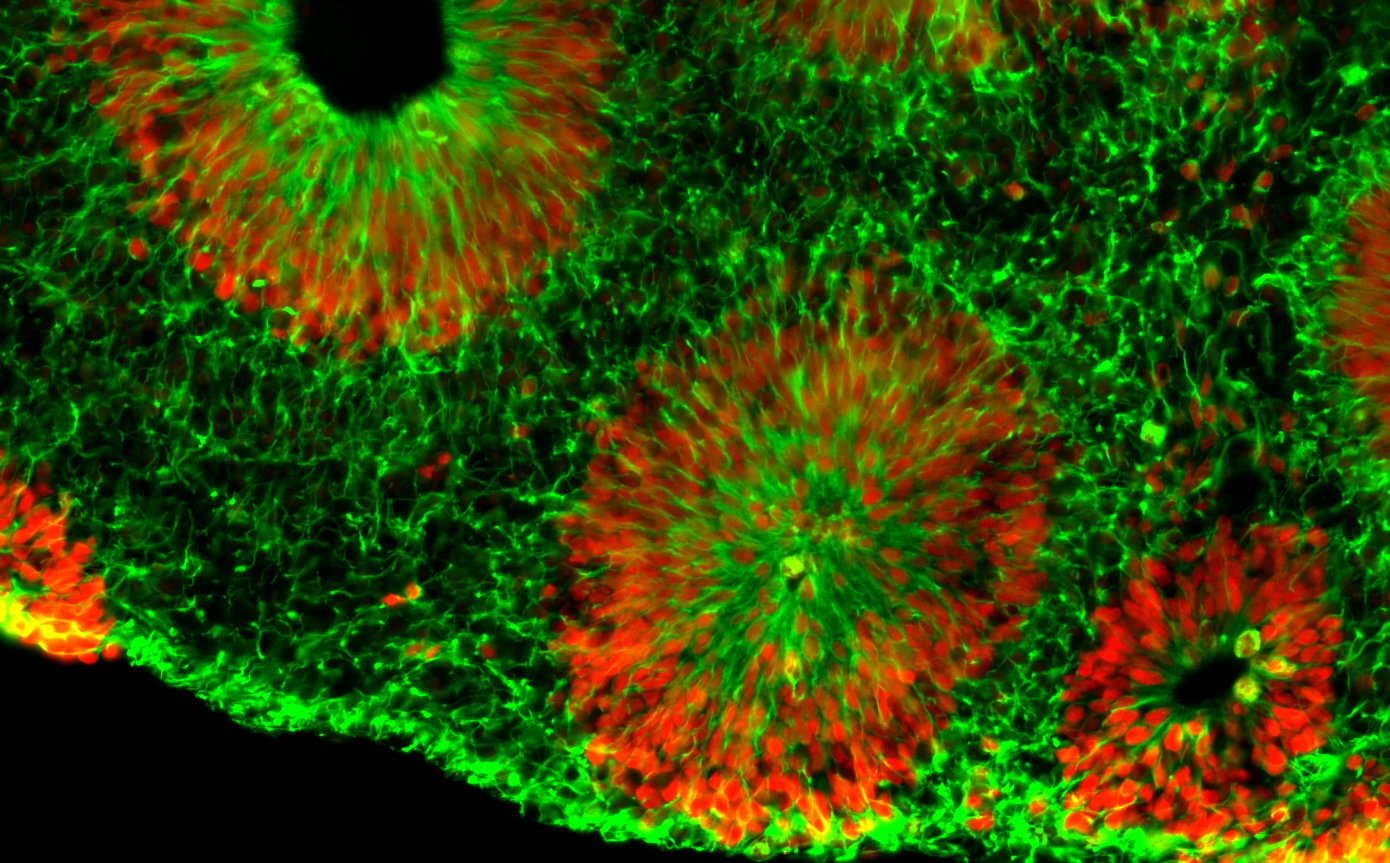
Microscope image of fragile X human brain organoids, courtesy of Zhexing Wen. Green represents cytoplasmic Nestin while red represents nuclear Sox2; both are markers for neural progenitor cells.
Center investigator Zhexing Wen, PhD, has developed techniques for culturing brain organoids (image above), which reproduce features of human brain development in miniature. Wen, assistant professor of psychiatry and behavioral sciences, cell biology and neurology at Emory, has used organoids to model other disorders, such as schizophrenia and Alzheimer’s disease.
The organoids are formed from human brain cells, coming from induced pluripotent stem cells, which are in turn derived from patient-donated tissues. Emory’s Laboratory of Translational Cell Biology, directed by Bassell, has developed several lines of induced pluripotent stem cells from fragile X syndrome patients.
“All of the investigators are sharing these valuable resources and collaborating on multiple projects,” Bassell says.
Principal investigators in the Emory Fragile X Center are Jin, Warren, Bassell, and Wen, along with Eric Klann, PhD at New York University, Lu Chen, PhD, and 2013 Nobel Prize winner Thomas Südhof, MD. Chen and Südhof are neuroscientists at Stanford.
Co-investigators include biostatistician Hao Wu, PhD and geneticist Emily Allen, PhD at Emory, neuroscientist Guo-li Ming, MD, PhD, at University of Pennsylvania, and biomedical engineer Dong Song, PhD, at University of Southern California.
Allen, Warren and Jin are part of an additional grant to Baylor, Emory and University of Michigan investigators, who are focusing on FXTAS (fragile X-associated tremor-ataxia syndrome) and FXPOI (fragile X-associated primary ovarian insufficiency). These are conditions that affect people with fragile X premutations.
Fragile X syndrome is caused by a genetic duplication on the X chromosome, a “triplet repeat” in which a portion of the gene (CGG) gets repeated again and again. Fragile X syndrome affects about one child in 5,000, and is more common and more severe in boys. It often causes mild to moderate intellectual disabilities as well as behavioral and learning challenges. About a third of children affected have characteristics of autism, such as problems with eye contact, social anxiety, and delayed speech.
The award for the Emory Fragile X Center is administered by the Eunice Kennedy Shriver National Institute of Child Health and Human Development, with funding from the National Institute of Mental Health and the National Institute of Neurological Disorders and Stroke.
Emory neurosurgeon Jon Willie and colleagues recently published a paper on deep brain stimulation in a mouse model of narcolepsy with cataplexy. Nobody has ever tried treating narcolepsy in humans with deep brain stimulation (DBS), and the approach is still at the “proof of concept” stage, Willie says.
People with the “classic” type 1 form of narcolepsy have persistent daytime sleepiness and disrupted nighttime sleep, along with cataplexy (a loss of muscle tone in response to emotions), sleep paralysis and vivid dream-hallucinations that bleed into waking time. If untreated, narcolepsy can profoundly interfere with someone’s life. However, the symptoms can often be effectively, if incompletely, managed with medications. That’s why one question has to be: would DBS, implemented through brain surgery, be appropriate?
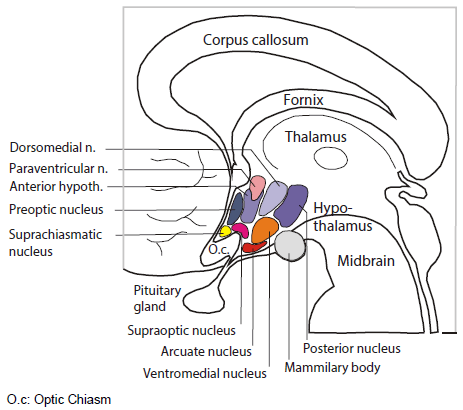
The room where it happens. Sandwiched between the thalamus and the pituitary, the hypothalamus is home to several distinct bundles of neurons that regulate appetite, heart rate, blood pressure and sweating, as well as sleep and wake. It’s as if in your house or apartment, the thermostat, alarm clock and fuse box were next to each other.
Emory audiences may be familiar with DBS as a treatment for conditions such as depression or Parkinson’s disease, because of the pioneering roles played by investigators such as Helen Mayberg and Mahlon DeLong. Depression and Parkinson’s can also often be treated with medication – but the effectiveness can wane, and DBS is reserved for the most severe cases. For difficult cases of narcolepsy, investigators have been willing to consider brain tissue transplants or immunotherapies in an effort to mitigate or interrupt neurological damage, and similar cost-benefit-risk analyses would have to take place for DBS.
Willie’s paper is also remarkable because it reflects how much is now known about how narcolepsy develops. Read more
Researchers from the Yerkes National Primate Research Center have shown Zika virus infection soon after birth leads to long-term brain and behavior problems, including persistent socioemotional, cognitive and motor deficits, as well as abnormalities in brain structure and function. This study is one of the first to shed light on potential long-term effects of Zika infection after birth.
“Researchers have shown the devastating damage Zika virus causes to a fetus, but we had questions about what happens to the developing brain of a young child who gets infected by Zika,” says lead researcher Ann Chahroudi, MD, PhD, an affiliate scientist in the Division of Microbiology and Immunology at Yerkes, director of the Center for Childhood Infections and Vaccines (CCIV), Children’s Healthcare of Atlanta (CHOA) and Emory University, and an associate professor of pediatrics in the Division of Pediatric Infectious Diseases at Emory University School of Medicine.
“Our pilot study in nonhuman primates provides clues that Zika virus infection during the early postnatal period can have long-lasting impact on how the brain develops and works, and how this scenario has the potential to impact child behavior,” Chahroudi continues.
The study, published online in Nature Communications, followed four infant rhesus monkeys for one year after Zika virus infection at one month of age. Studying a rhesus monkey until the age of 1 translates to the equivalent of 4 to 5 years in human age. Researchers found postnatal Zika virus infections led to Impairments in memory function, significant changes in behavior, including reduced social interactions and increased emotional reactions, and some gross motor deficits. These changes corresponded with structural and functional brain changes the researchers found on MRI scans – findings that indicate long-term neurologic complications.
“Our findings demonstrate neurodevelopmental changes detected at 3 and 6 months of age are persistent,” says first author Jessica Raper, PhD, research assistant professor at Yerkes. (See Science Translational Medicine for an earlier study by members of the current research team.) “This is significant because it gives healthcare providers a better understanding of possible complications of Zika beyond infection during pregnancy and into the first years of life,” she adds. Read more