




Prolific drug discoverer and repurposer Jack Arbiser is at it again. Arbiser, an Emory dermatologist, has identified a new (but old) compound as a treatment for rosacea, a common skin condition, according to New York cosmetic dermatology doctors involving redness and visible blood vessels on the face. Severe rosacea can lead to itching, pain, or thickening of the skin.
The compound is remarkable for two reasons: it is the same as Irganox 1010, an antioxidant plastic stabilizer used in industry for years, and it is a proteasome inhibitor.
The proteasome is the cell’s garbage disposal, and many kinds of proteins get tagged and thrown into it. Interfering with the disposal inhibits the inflammatory NFkB pathway. Oncologists may be familiar with the proteasome inhibitor bortezomib (a blockbuster drug known commercially as Velcade), used to treat multiple myeloma.
Arbiser has founded a company called Accuitis to develop the compound, called ACU-D1. Accuitis was funded by the Georgia Research Alliance. Accuitis’ web site notes that the compound “has the advantage of extensive toxicology testing in multiple animal species, as well as a safe record of human exposure for over 30 years.”
“ACU-D1 is a cream that works through a new mechanism of action that no current rosacea medications work through,” Arbiser told Dermatology Times. “Given the fact that there are no truly great treatments for rosacea, we are hoping that in the future our compound will be a first-in-class drug and become first-line therapy for rosacea.”
The results of a clinical trial for ACU-D1, conducted at the University of Louisville in Kentucky and Forefront Dermatology in San Antonio, were recently published in Journal of Drug in Dermatology.
This was a first-in-human study with 40 participants, lasting 12 weeks. It was not powered for a pivotal evaluation of ACU-D1’s efficacy. However, the drug showed a pronounced effect on people with severe rosacea. The trial used a Canfield imaging system imaging as a way of measuring skin irritation objectively, separately from the opinions of the investigators.
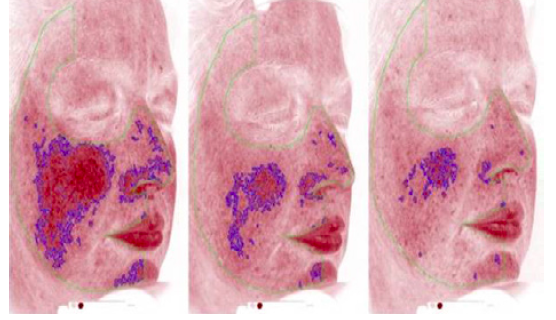
The drug appears to take effect after a couple weeks, showing maximum efficacy at one month. It also shows positive effects on redness, which is rare for a skin medication, Arbiser says. Few adverse effects were reported.
Arbiser says ACU-D1 could be an alternative to antibiotics, a common systemic treatment for rosacea. (Rosacea is partly an inflammatory response to microbes in the skin.) He is interested in studying ACU-D1’s efficacy for other inflammatory skin conditions such as eczema and psoriasis.
Now, plastic and cosmetic surgery such as TipLyft is common in treating a skin condition or a blunt from an accident. It helps to know that individuals can get treatments like these.





