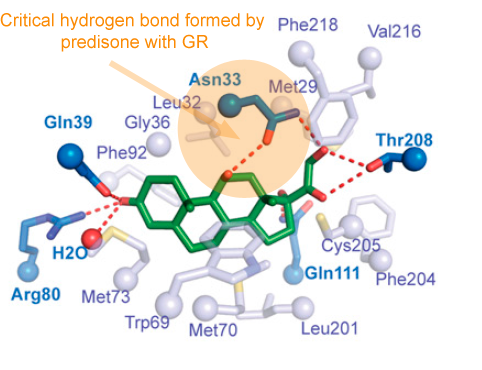
How many people out there have been exposed to SARS-CoV-2? It’s a tricky question, once you think about all the people who have experienced COVID-19 symptoms over the last several months, but didn’t go to the hospital. And there’s a murkier penumbra of people who may have fended off the virus with a minor immune skirmish.
A recent Emerging Infectious Diseases paper from Emory investigators includes antibody tests on a group of more than 100 adults in the Atlanta area who experienced mild flu-like symptoms this spring, but couldn’t get tested for SARS-CoV-2 itself.
A sizable fraction (22 to 48 percent, depending on when they provided blood samples) had elevated levels of IgM against the coronavirus. IgM is the “rookie” antibody produced when the immune system is first encountering something, as opposed to the more seasoned IgG, which appears later in an immune response and tended to rise only in people who were hospitalized. The Emory authors came to a conclusion that others are also reaching:
“Examining IgM and IgG against multiple SARS-CoV-2–related antigens may thus better inform natural history and vaccine studies than any one antibody.”
To answer these kinds of questions more comprehensively, investigators will need to go broader. For example, this week the American Red Cross published data on what proportion of its blood donors have antibodies against SARS-CoV-2. About 3 percent of first-time donors did, using their criteria.
For big answers, we can look to studies such as Emory’s COVID-Vu, a nationwide population-based study using antibody and virus tests taken at home. Rollins School of Public Health researchers received a $6.6 million grant to launch the study this summer. This type of study is designed to cover everyone, whether they were sick or not.